

/image%2F2722116%2F20180323%2Fob_f2782d_aidonsnosenfantsaretrouverconfianceen.jpg)
Démêler la tauopathie de la maladie d’Alzheimer et du parkinsonisme[1]
Sclérose en plaques. Cerveau et cancer.
Résumé :
Le Tau est une protéine associée aux microtubules qui se localise principalement dans l'axone pour stabiliser la structure des microtubules axonaux et la connectivité neuronale. La pathologie du tau est l’une des protéinopathies les plus courantes associée aux maladies neurodégénératives dépendantes de l’âge, notamment la maladie d’Alzheimer et divers parkinsonismes. La protéine Tau subit une pléthore de modifications intramoléculaires et certaines formes modifiées favorisent la production de Tau oligomère toxique et de filaments hélicoïdaux appariés, à travers lesquels ils s'assemblent en outre dans des enchevêtrements neurofibrillaires, également appelés tauopathie. Dans cette revue, nous discuterons des avancées récentes de la recherche sur la tauopathie, en mettant l'accent sur son association avec la manifestation axonale précoce du défaut de transport axonal, le stress mitochondrial axonal, l'accumulation de vésicules autophagiques et le processus de destruction d'axones, et la propagation pathogène du Tau à travers la synapse. Deux stratégies alternatives, soit en ciblant la protéine Tau elle-même, soit en améliorant le déclin physiologique lié à l'âge, sont actuellement en cours de réalisation pour trouver le traitement prometteur contre la tauopathie. Il ne fait aucun doute que des études supplémentaires sont nécessaires pour lutter contre cette maladie dévastatrice qui a déjà touché des millions de personnes dans notre population vieillissante.
Contexte
On peut ou non développer une lésion de tauopathie, même lorsque les membres de sa famille sont atteints de la maladie d’Alzheimer ou de la maladie de Parkinson - cela semble être une bonne nouvelle car la plupart des cas de tauopathie sont sporadiques. Cependant, c'est aussi un sentiment qui montre notre manque de compréhension de cette protéinopathie couramment observée dans les cerveaux vieillissants. Le légendaire psychiatre allemand Alois Alzheimer a été le premier à avoir décrit la tauopathie. Il a vu son patient âgé de 51 ans, Auguste, perturbé par des symptômes de démence, notamment une perte de mémoire à court terme, une dépression et un délire. Alzheimer a découvert que le cerveau post-mortem d’Auguste contenait des enchevêtrements neurofibrillaires et des plaques séniles, ces protéinopathies plus tard considérées comme le signe distinctif de la pathologie de la MA (maladie d’Alzheimer). Nous savons aujourd'hui que les protéines Tau forment cette structure ressemblant à des fibrilles. En fait, diverses formes d'agrégation de Tau représentent plus de 20 troubles neurologiques, dont la MA, la démence frontotemporale avec parkinsonisme-17 (FTDP-17), la paralysie supranucléaire progressive, la dégénérescence corticobasale, la maladie du grain argyrophilique, l'encéphalopathie traumatique chronique et la maladie de Parkinson (MP). Quelle est l'étiologie de cette pathologie dévastatrice développée et comment affecte-t-elle notre fonctionnement cérébral ? Dans cette revue, nous résumons les progrès récents dans la recherche sur la tauopathie et mettons en évidence certains mécanismes physiopathologiques qui pourraient évidemment causer des dommages à notre cerveau au cours du processus de vieillissement.
Connectivité neuronale
La pathologie du Tau transmise par la CE (cortex entorhinal) pourrait provoquer des déficits dans l'activation des éléments de réseau et perturber la cognition spatiale chez la souris, comme ce fut le cas lors de la maladie d'Alzheimer (MA). En effet, on pense que l'accumulation pathologique de tauopathies dans la CE est la première synapse où les germes de Tau influencent les circuits de la mémoire dans le cerveau, et cette évolution pathologique est associée à une perte progressive de la mémoire épisodique dans la MA (Fig. 1).

Fig. 1 Un dessin schématique résume les mécanismes pathogènes possibles dans la tauopathie précoce et sa propagation ultérieure. La protéine Tau peut rencontrer différentes modifications post-traductionnelles, et certaines modifications pourraient conduire à la formation d'oligomères de Tau et au développement subséquent de la structure de classe supérieure, telle que le filament à paires hélicoïdales présenté dans l'image EM. La fonction Tau modifiée peut entraîner une accumulation aberrante des mitochondries, un stress oxydatif, des lésions de l'ADNmt et une altération de l'autophagie de l'axone. Ces conditions endommagent l'homéostasie axonale et peuvent permettre aux Tau pathogènes de se propager à la cellule post-synaptique. Nous avons intégré les connaissances actuelles sur la propagation du tau et ses liens potentiels avec les mécanismes de sécrétion. Dans des conditions physiologiques, la sécrétion de Tau dépend de l'activité (23). Nous avons incorporé les connaissances actuelles sur la propagation du tau et ses liens potentiels avec les mécanismes de sécrétion connexes: le tau non gardé (66); exosome (108); exophore (109).
Protéine Tau associée aux microtubules
Dans des conditions physiologiques, Tau existe à l'état non plié et 80% des protéines Tau interagissent avec les microtubules des axones Lorsque Tau n'interagit pas avec d'autres protéines, il peut s'envelopper tout seul et cet état de liaison intramoléculaire aléatoire est jugé nécessaire pour prévenir les interactions avec une protéine Tau supplémentaire en masquant les répétitions de liaison aux microtubules. La protéine elle-même est bipolaire ; le domaine N-terminal est fortement chargé négativement dans un point isoélectrique de 3,8, alors que les répétitions du domaine riche en proline et de la liaison aux microtubules sont chargées positivement dans un point de charge isoélectrique de 11,4 et 10,8 respectivement. [Rappelons ici qu’un polymère "amine" dérivé des polymères poly(L-lysine) est un semi-conducteur.[2]] Il semble que des modifications post-traductionnelles pourraient modifier la charge de Tau et son état de liaison. La phosphorylation de certains résidus dans le domaine de liaison aux microtubules peut neutraliser la charge positive et dissocier la protéine tau des microtubules. Le Tau est très hydrophile et ne contient qu'une petite partie des résidus hydrophobes. En outre, la protéine Tau normale ne présente pas de structure secondaire, si elle n’est que transitoire, et l’interaction de liaison des résidus Tau chargés positivement et des résidus chargés négativement dans les dimères de microtubules ou de tubuline pourrait former une structure hélicoïdale dans le domaine de liaison aux microtubules de Tau.
Il a été démontré que Tau protégeait l'ADN dans le noyau.
L'inactivation de la protéine Tau chez la souris pourrait entraîner un transport axonal défectueux de la protéine précurseur de l'amyloïde (APP), une accumulation de fer intraneuronal et une perte neuronale dans la substance noire. De plus, les cerveaux dépourvus de protéine Tau pourraient améliorer les sphéroïdes axonales anormales dans les voies de la substance blanche chez un modèle murin de souris APP humain muté pour la MA. Il a également été suggéré que la régulation temporelle et spatiale précise des partenaires de liaison à la protéine Tau permet aux neurones de contrôler les fonctions de la protéine Tau, ce qui est essentiel pour la fonction et la plasticité des neurones.
Mutations Tau dans les maladies neurologiques humaines
La Tau est l'une des protéines les plus couramment impliquées dans différentes maladies neurodégénératives. Nous utilisons aujourd'hui la tauopathie pour trouver des conditions neurologiques impliquant le rôle pathogène d'agrégats de Tau. On estime que plus de 30 millions de personnes souffrent actuellement de maladies neurologiques associées à la tauopathie, notamment la MA, la MP, le FTDP-17, le complexe parkinsonisme-démence de Guam et la dégénérescence corticobasale, la paralysie supranucléaire progressive, le syndrome corticobasal, et l’encéphalopathie traumatique. Des mutations du gène Tau ont été découvertes dans la forme bien connue de démence fronto-temporale (FTD), un trouble générique couvrant une gamme de conditions cliniques telles que la maladie de Pick, la démence corticobasale et la paralysie supranucléaire progressive.
Comment les protéines Tau naturellement dépliées peuvent-elles s'agréger en filaments tachés hélicoïdaux et droits parfaitement appariés périodiques ? Les analyses atomiques à haute résolution ont récemment affiné la structure des noyaux de filaments. Le modèle actuel basé sur la structure Cryo-EM de filaments de tau montre que deux protofilaments jumeaux comprenant environ 70 acides aminés (résidus 306 à 378, correspondant à R3 et R4 des protéines tau) pourraient former une structure croisée en hélice bêta/bêta.
Modifications post-traductionnelles de Tau
Les patients atteints de tauopathies portent rarement le gène MAPT muté. Ainsi, la manière dont les protéines Tau normales peuvent devenir dysfonctionnelles et s'auto-assembler dans la pathologie Tau est un mystère. Ces modifications de Tau seraient à l'origine de la pathogenèse de Tau et constituent depuis des années le domaine central de la recherche sur Tau. Les modifications post-traductionnelles de Tau pourraient entraîner la perte de la structure non pliée native de la protéine, et favoriser ainsi la formation de la forme de plaque C bêta pour déclencher l'agrégation de Tau.
a. Phosphorylation / Déphosphorylation
Comme beaucoup de substrats de kinase-phosphatase, Tau peut être phosphorylé par des kinases et leur phosphorylation peut être inversée par une déphosphorylation via des phosphatases. Les protéines phosphatases de la protéine tau ont été identifiées comme PP2A, PP1, PP2B, PP2C et PP5. Le Tau hyperphosphorylé pourrait être dû à la diminution d'environ 20–40% de l'activité de PP2A, ce qui montre que le lien entre les kinases et les phosphatases contrôle la propriété de Tau. Bien que l’opinion traditionnelle considère que l’hyperphosphorylation de Tau est toxique pour le neurone affecté en raison de la nature de la pathologie cérébrale de la MA, cependant, inverser l'hyperphosphorylation de Tau en soi peut ne pas suffire à guérir la neurodégénérescence, car l'inhibition de la kinase était incapable de bloquer la lésion induite par Tau.
b. Troncature
Certaines troncatures de Tau pourraient empêcher sa nature dépliée et favoriser ainsi l'agrégation de Tau.
c. Acétylation/désacétylation
L'autre strate de la modification de la protéine Tau est l'acétylation et la désacétylation. Tau a 23 résidus de lysine et 13 sites de lysine se trouvent dans le domaine de liaison aux microtubules, qui comprend KXGS préservé dans les quatre répétitions. Des enzymes telles que la p300 acétyltransférase et la sirtuine dépendante de NAD+, dont la fonction est la désacétylase, pourraient agir sur Tau. Une étude pionnière a suggéré que le manque d'acétylation de Tau pouvait être associé à la maladie du grain argyrophilic (AGD), une condition de démence légère. Comme les cerveaux de AGD présentaient une pathologie réduite de la protéine Tau, il était donc possible que la prévention de l'acétylation de la protéine Tau dans notre cerveau soit un mécanisme de défense essentiel contre la propagation d'agrégats de tau. Paradoxalement, il a été rapporté que le motif d'acétylation KXGS susmentionné est hautement ubiquitiné dans le cerveau MA humain], indiquant qu'une acétylation de la protéine tau dans le domaine de liaison aux microtubules pourrait empêcher sa dégradation. Ainsi, l'ubiquitination et l'acétylation des mêmes résidus de lysine dans le Tau peuvent créer un état complexe pour le renouvellement du Tau.
Existence de suppresseurs moléculaires dans le cerveau et leur importance dans les défauts cognitifs associés au vieillissement, l’Alzheimer et la neurodégénérescence[3] : deux phosphatases, nommées la protéine phosphatase 1 (PP1) et la calcineurine sont des catalyseurs de l’oubli et de la perte des facultés cognitives chez la souris
Le Prof. Isabelle Mansuy et son équipe s’intéressent en particulier aux bases génétiques et épigénétiques des fonctions cognitives. Alors que de nombreuses équipes étudient le rôle d’un groupe d’enzymes bien connues, les kinases, Isabelle Mansuy s’est-elle, très tôt, tournée vers des enzymes quasi méconnues, les phosphatases. Ses travaux ont démontré que deux phosphatases, nommées la protéine phosphatase 1 (PP1) et la calcineurine sont des catalyseurs de l’oubli et de la perte des facultés cognitives chez la souris. De tels troubles de la mémoire sont généralement associés à l’âge et se manifestent particulièrement chez les individus souffrant de Morbus Alzheimer ou de neurodégénération. « Quand la production de PP1 ou de calcineurine est réprimée par manipulation génétique, nous observons que des souris d’âge mûr ont une mémoire aussi fonctionnelle que celle de jeunes souris », rapporte Isabelle Mansuy. Une de ces protéines agit comme un régulateur épigénétique de gènes nécessaires à la formation de la mémoire.
L’environnement a une influence capitale sur notre vie : certains événements peuvent laisser des marques biochimiques sur notre ADN pour toute la vie. De telles marques peuvent s’imprimer dans toutes les cellules du corps y compris dans le cerveau. Isabelle Mansuy, professeur de neuro-épigénétique à la faculté de médecine de Zurich et à l’école polytechnique fédérale de Zurich, est une pionnière dans le domaine de la neuro-épigénétique, un domaine de recherche qui étudie l’influence de l’environnement sur le cerveau. Ses découvertes scientifiques contribuent, de manière déterminante, à la compréhension des maladies cognitives et psychiatriques. Ses recherches ont démontré l’existence de suppresseurs moléculaires dans le cerveau et leur importance dans les défauts cognitifs associés au vieillissement, l’Alzheimer et la neurodégénérescence.
Isabelle Mansuy a étudié la biologie et la biotechnologie à l’Université Louis Pasteur de Strasbourg. Elle a obtenu un doctorat en neurobiologie également à l’Université Louis Pasteur de Strasbourg pour un travail de recherche effectué à l’institut Friedrich Miescher à Bâle. De 1994 à 1998, elle était postdocteur à l’Université Columbia à New York puis a obtenu un poste de Professeur assistant à l’EPFZ en Décembre 1998. Elle a été nommée professeur ordinaire à l’Université de Zurich et à l’EPFZ (chaire double) en 2012. Elle a reçu la Médaille du Chevalier dans l’Ordre National du Mérite en France.
Le Prof. Isabelle Mansuy et son équipe s’intéressent en particulier aux bases génétiques et épigénétiques des fonctions cognitives et des réponses comportementales chez les mammifères. Alors que de nombreuses équipes étudient le rôle d’un groupe d’enzymes bien connues, les kinases, Isabelle Mansuy s’est-elle, très tôt, tournée vers des enzymes quasi méconnues, les phosphatases. Ses travaux ont démontré que deux phosphatases, nommées la protéine phosphatase 1 (PP1) et la calcineurine sont des catalyseurs de l’oubli et de la perte des facultés cognitives chez la souris. De tels troubles de la mémoire sont généralement associés à l’âge et se manifestent particulièrement chez les individus souffrant de Morbus Alzheimer ou de neurodégénération. « Quand la production de PP1 ou de calcineurine est réprimée par manipulation génétique, nous observons que des souris d’âge mûr ont une mémoire aussi fonctionnelle que celle de jeunes souris », rapporte Isabelle Mansuy. Une de ces protéines agit comme un régulateur épigénétique de gènes nécessaires à la formation de la mémoire. Est-ce que ces protéines ont la même fonction chez l’humain que chez la souris ? Ceci reste encore à prouver. Si c’est le cas, il s’agirait là d’un point de départ possible pour le développement de nouvelles thérapies de traitement des troubles de la mémoire.
Il est connu depuis longtemps en clinique, que certains troubles du comportement, en particulier, ceux dus à des événements traumatiques vécus pendant l’enfance ou l’adolescence, peuvent être transmis à travers les générations. Ce qui reste encore méconnu cependant sont les mécanismes responsables de ces troubles et de leur transmission. De nouvelles données récentes ont apporté des éléments inattendus à cette question: « Nous avons pu montrer, chez un modèle de souris, que les conséquences de traumatismes précoces sur le comportement peuvent se manifester jusqu’à deux générations plus tard, au niveau des petits-enfants, bien que ceux-ci n’aient subi aucun traumatisme. », explique Isabelle Mansuy. Non seulement le comportement des souris est altéré mais des anormalités au niveau du métabolisme sont également observées. « Ces changements sont en partie dus à un déséquilibre au niveau des micro-ARNs. », relève Isabelle Mansuy. Les micro-ARNs sont de petits fragments d’ARN ayant pour fonction la régulation de nos gènes. Isabelle et son équipe étudient actuellement les mécanismes responsables de leur déséquilibre dans le cerveau et les cellules germinales. « Comme nous savons que des micro-ARNs présents dans le sperme sont responsables de la transmission des effets des traumatismes et que les même micro-ARNs sont altérés dans le sérum chez la souris, nous espérons découvrir un biomarqueur, présent dans le sang des patients, qui permettrait de diagnostiquer de telles maladies », précise Isabelle Mansuy.[4]
Effet de l’hydroxycitrate (ou acide hydroxycitrique), un dérivé de l’acide citrique, sur la sclérose en plaques
De nombreuses études plus anciennes ont souligné l’action anti-inflammatoire de l’acide hydroxycitrique, qui est en fait un acide-alcool ou acide de fruit, dérivé de l’acide citrique, une substance naturellement présente dans le citron et l’orange qui leur confère un goût aigre. En se basant sur cette hypothèse, des chercheurs ont utilisé cette molécule chez des modèles murins souffrant de sclérose en plaques afin d’évaluer son efficacité. Cette maladie auto-immune, rappelons-le, peut provoquer une démyélinisation (détérioration de la myéline qui est la gaine d’isolation des cellules nerveuses) et une altération de l’axone (partie allongée d’un neurone qui assure le passage des informations). Ce qui conduit à une perturbation de nombreux processus biologiques, dont l’immunité et l’inflammation.

Pendant 3 semaines, les sujets expérimentés ont reçu 2g/kg/j d’acide hydroxycitrique. Les résultats du contrôle réalisé au terme du traitement ont montré une amélioration des parties lésées causées par la sclérose en plaques. Cet acide de fruit a agi à la manière d’un neuroprotecteur. Il a, par ailleurs, limiter les effets néfastes des espèces réactives de l’oxygène (malondialdéhyde, oxyde nitrique) dues aux stress oxydatif, et ceux de l’inflammation chronique, en inhibant l’action des médiateurs inflammatoires.[5]
Comment le cerveau participe au cancer[6] : les dernières avancées
Une équipe de biologistes vient de découvrir que le cerveau produit des cellules souches neuronales qui, via les vaisseaux sanguins, vont infiltrer des tumeurs cancéreuses en formation. Cette découverte ouvre une piste thérapeutique.[7]
Quelle est la découverte des neurobiologistes français ?
Une équipe de biologistes du laboratoire cancer et micro-environnement (Inserm-CEA) à Fontenay-aux-Roses vient de découvrir que le cerveau produit des cellules souches neuronales qui, franchissant la barrière hématoencéphalique – l’enveloppe du cerveau pourtant réputée infranchissable – vont être transportées par le sang et vont aller infiltrer des tumeurs cancéreuses en formation, notamment dans la prostate.
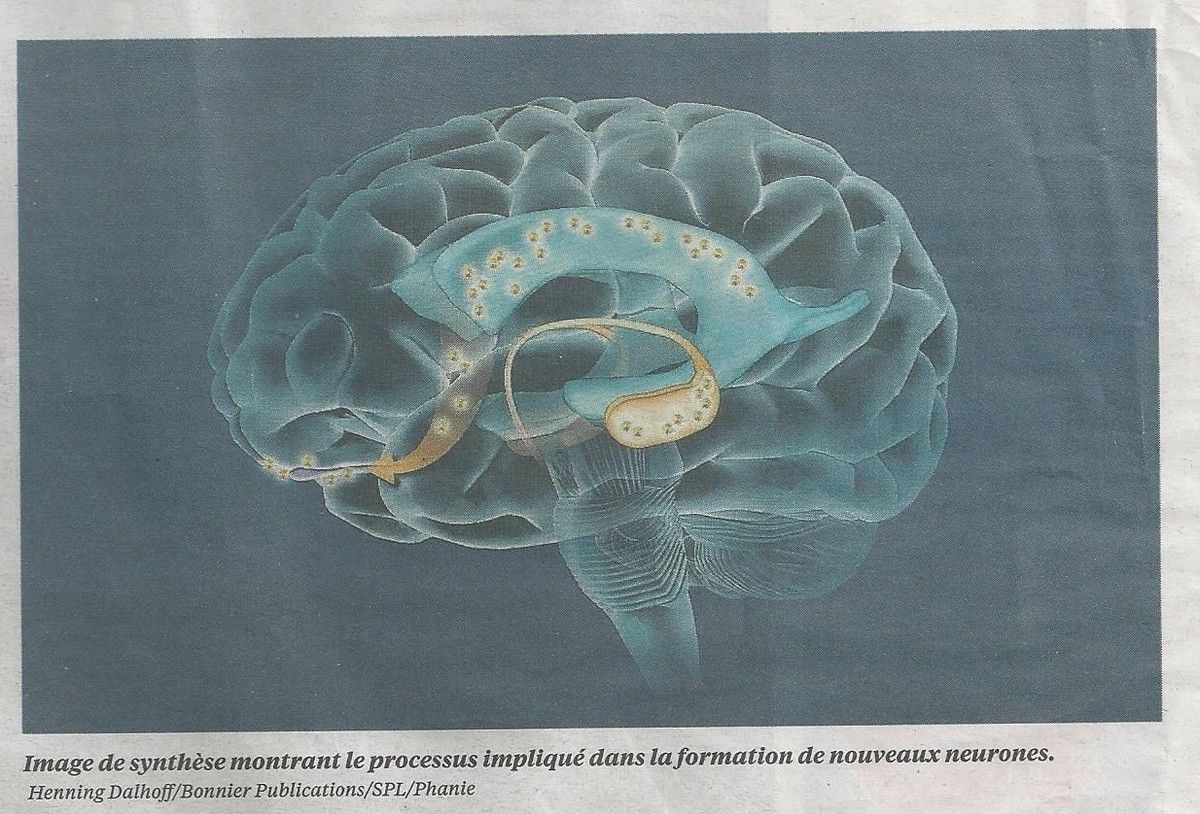
Cette découverte ouvre la voie à un nouveau champ de recherche, relatif au rôle du système nerveux dans le développement des cancers et aux interactions si importantes entre les systèmes vasculaires, immunitaires et nerveux dans la formation d’une tumeur.
Comment les chercheurs ont-ils procédé ?
Déjà en 2013, cette équipe de biologistes menée par Claire Magnon avait mis en évidence, dans des tumeurs de la prostate que l’infiltration de fibres nerveuses était associée à la survenue et à la progression de ce cancer. Depuis, d’autres études ont permis de confirmer le rôle inattendu, mais apparemment important, des fibres nerveuses dans le micro-environnement tumoral de nombreux cancers solides (poumon, sein).
En étudiant les tumeurs de 52 patients atteints de cancer de la prostate, claire Magnon y a découvert des cellules particulières, appelées « cellules progénitrices neuronales ». Ces cellules, normalement, ne s’observent que lors du développement embryonnaire ou chez l’adulte, et ne se trouvent que dans deux zones du cerveau, l’hippocampe et la zone sous-ventriculaire. De plus, le nombre de ces « cellules vagabondes » est parfaitement corrélé à la sévérité du cancer.
Pour déterminer l’origine de ces cellules progénitrices neuronales, les chercheurs ont utilisé des souris transgéniques, porteuses de tumeurs. Ils ont pu démontrer que, lors de l’établissement d’une tumeur, les cellules nichées dans la zone sous-ventriculaire passaient dans la circulation sanguine. Cette migration s’accompagne d’anomalies de perméabilité de la barrière hématoencéphalopatique.
« Pour l’instant, rien ne permet de savoir si ce problème de perméabilité précède l’apparition du cancer, ou si elle est provoquée par le cancer lui-même via des signaux issus de la tumeur en formation. Quoi qu’il en soit, ces cellules migrent dans le sang jusqu’à la tumeur où elles s’intègrent au micro-environnement. Là, elles se spécialisent en neurones produisant un neurotransmetteur, l’adrénaline. Or, l’adrénaline régule le fonctionnement du système vasculaire, et c’est probablement ce mécanisme qui favorise à son tour le développement tumoral. Mais ces hypothèses restent à vérifier », conclut prudemment la chercheuse.
Quelle piste de traitement ouvre cette découverte ?
Cette recherche ouvre la porte à une nouvelle piste thérapeutique. Des observations cliniques montrent déjà que les patients atteints de cancer de la prostate qui utilisent des bêtabloquants (substance qui annihile les effets négatifs d’un taux d’adrénaline chroniquement élevé en bloquant les récepteurs de l’adrénaline) à des fins cardiovasculaires, présentent de meilleurs taux de survie. « Il serait intéressant de tester ces médicaments en tant qu’anticancéreux », estime la chercheuse. Deux essais cliniques ont commencé aux États-Unis.
[3] https://radionotredame.net/emissions/enquetedesens/21-05-2019/
[4] https://www.gensuisse.ch/fr/blog/prof-isabelle-mansuy-facult%C3%A9-de-m%C3%A9decine-universit%C3%A9-de-z%C3%BCrich-et-d%C3%A9partement-des-sciences-et
[5] Mahdi G et al., L’acide hydroxycitrique améliore l’inflammation et le stress oxydatif chez les modèles murins de sclérose en plaques, recherche sur la régénération neurologique.
https://www.santescience.fr/hydroxycitrate/
[6] DENIS SERGENT, Comment le cerveau participe au cancer, article du 17 mai 2019, La Croix.
[7] Publié le 15 mai 2019 dans Nature.